Unraveling multi-electron dynamics is important, as it initiates the electron transfer that is often concerted with bond breaking and bond formation. Therefore, it plays a pivotal role in chemistry and biology by enabling control of photochemical processes like energy conversion. It is also important for emerging technologies like nano- and bio-photonics (sensing, probing, and nano-plasmonic devices), and molecular electronics as it determines the efficiency with which energy is coupled thereby enabling spatial localization.
Ultrafast dynamics in complex molecules
-
Imaging and controlling electrons on their natural time scale is one of the greatest challenges in science for the 21st century. Until recently, time resolving such phenomena have been out of the reach of science. However, research on attosecond technology over the last decade has changed the scientific landscape. It has spawned a new source of extreme ultraviolet (XUV) radiation producing the shortest optical pulses and enabled us to image molecular orbitals and to measure position of the atoms in molecules. Attosecond technology is based on high harmonics generation (HHG) – XUV light emitted when an electron, removed from an atom by an intense laser field, is driven back to recombine with the parent ion.
-
Attosecond pump – probe spectroscopy is the most direct method to probe ultrafast electron dynamics; however, experiments are complex and challenging. In a second approach, ultrafast dynamics is initiated by an intense fs pulse (or as pulse) and probed by an as pulse (or fs pulse). A third approach involves ionization by an intense infrared fs pulse and probing by recollision, which results in the emission of XUV photons leading to HHG spectroscopy. Our research primarily employs the third approach with the specific goal of studying ultrafast processes, specifically multi-electron dynamics, in complex molecules.
Various Research Projects
High harmonic spectroscopy of complex molecules
-
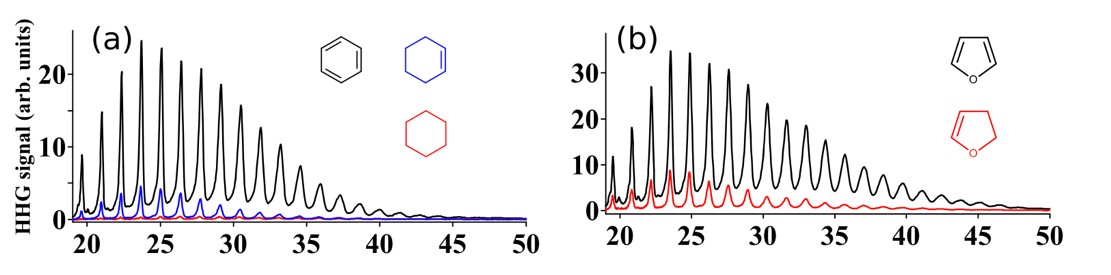
The amplitude and phase of the generated harmonics can be used as a sensitive tool to probe electronic structure and nuclear dynamics with Angstrom spatial and as temporal precision. However, deciphering electronic and molecular structure information from HHG, both experimentally and theoretically, has been possible only in simple aligned molecules. Multi-electron dynamics and complex electronic structure of polyatomic molecules makes it difficult to extend HHG as a precision spectroscopic tool. We showed that fingerprints of symmetry and electronic structure could be decoded from HHG even in complex randomly oriented molecules. We demonstrated that HHG spectroscopy could be used to probe electron localization and to differentiate stereoisomers of randomly oriented molecules.
-
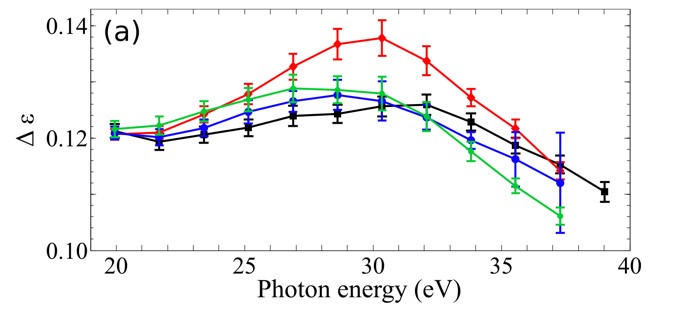
Recently, we introduced HHG spectroscopy as a new tool to probe aromaticity by demonstrating its capability in 5–membered ring molecules. Aromaticity is a manifestation of electron delocalization in cyclic molecules. It is a widely used qualitative concept in chemistry to understand behavior of molecules but there are no direct measures to quantify the degree of aromaticity. Presence of competing effects beside electron delocalization often results in disagreements among the different computational and experimental approaches. We extend these studies to larger ring structures so that HHG can be used as a generalized measure of aromaticity.
-
Identifying structural isomers (molecules with the same formula but different spatial arrangement of atoms) is a key task in analytical chemistry. Common techniques, such as mass spectrometry cannot distinguish between isomers as they have nearly identical electron impact and photoionization crosssections. Previously, we showed that HHG spectroscopy can distinguish stereoisomers. We extend our studies to three sets of structural isomers: (i) ortho-, meta- and para- xylenes in which the two methyl groups are attached differently to the carbon atoms (ii) ortho-, meta-, para-benzenediol in which two hydroxyl groups are substituted onto a benzene ring; and (iii) pyridine, pyrimidine, pyrazine and pyridazine that differ in the location of nitrogen atoms in the ring. This will provide insights into how the returning electron wavepacket in HHG perceives changes in the local environment within a molecule and enables the study of fast structural changes during isomerization.
Probing electron dynamics in complex molecules
-
An electron removed instantaneously from a complex molecule leads to two interesting processes; both play an important role in chemical and biological processes, and evolve on an as time scale: (i) The hole left behind moves back and forth from one end of the molecule to the other. Such a charge migration process is the initiator of photosynthesis, catalysis and DNA damage by ionizing radiation. (ii) Multi-electron excited states (MEES) are populated. These are bound states that lie below the ionization potential and promote energy conversion transformations by coupling electronic and nuclear degrees of freedom. An in-depth knowledge of these fundamental processes in nature is crucial, but very little has been explored to-date due to experimental and theoretical difficulties.
-
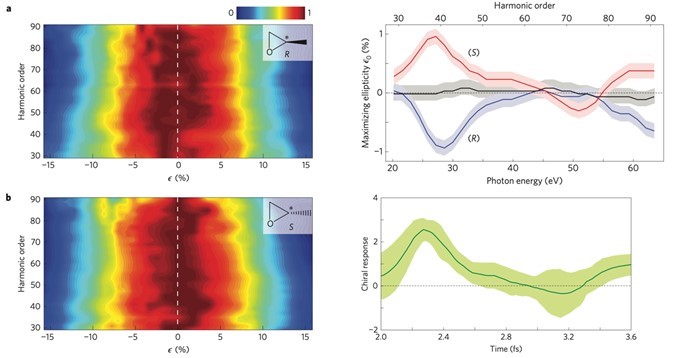
Using HHG spectroscopy, we showed that the laser field induced transitions between the ground and excited state of a molecular ion leads to as hole dynamics that are sensitive to molecular chirality. We demonstrated, chiral sensitivity of 3-10% in randomly oriented epoxypropane and fenchone, and probed the dynamics using a single color elliptically polarized laser field.
-
We use multi-color fields, such as the combination of a fundamental field and its second harmonic, to control the hole dynamics and therefore further enhance chiral sensitivity. This technique allows us to couple to specific orbitals/electronic states of the ion with appropriate properties even when ionization by a single color field does not involve favorable multi-orbital transitions. It also allows us to probe the dynamics by varying the relative phase between the two-color fields. We extend our studies to other chiral molecules such as alanine (simplest amino acid linked to hypertension and diabetes), uracil (one of the four nucleobases of RNA) and thymine (one of the four nucleobases of DNA).
Probing ring opening and isomerization dynamics
-
Nature harvests energy by using conjugation to control and direct chemical processes. For example, the photo-induced vision process and the formation of pre-vitamin D from 7-dehydrocholesterol take advantage of two interesting photochemical reactions: electrocyclic (rearrangement of π and σ bonds) leading to opening and closing of organic ring structures, and cis-trans isomerization. Both reactions occur on several 10’s of fs but proceed through a multitude of excited states whose dynamics occur on much faster time scales and are difficult to probe. In spite of 5 decades of studies little has been learned about the time-evolving electronic or vibrational states or about the molecular structure as it evolves in time.
-
In a pump-probe scheme, we initiate the photochemical reaction by a UV pump and probe the dynamics by HHG with an intense infrared pulse in the excited system. Using a transient grating setup for background-free measurement we monitor changes to the harmonic spectra and their phase. Our scheme allows us to study dynamics on two different time scales: structural evolution by varying pump-probe fs delay, and excited state evolution with sub-fs resolution – similar to what we observed recently in chiral molecules using the inherent temporal resolution of HHG spectroscopy. As an example, we can study the ring opening reaction of 1,3-cyclohexadiene (CHD) to form cis-1,3,5- hexatriene since it is analogous to the formation of previtamin D from 7-dehydrocholesterol.
Chiral Interactions
-
Left- and right-handed forms of a chiral molecule are indistinguishable unless they interact with asymmetric/chiral systems such as circularly polarized light. Optical excitation of chiral molecules results in (a) changes to the refractive index of the medium called optical activity/rotation, and (b) differential absorption of circularly polarized light called circular dichroism (CD). CD spectroscopy is the most widely used analytical method for conformational and structural characterization of biomolecules, and even magnetic properties of materials in X-ray regime. The main drawback is that it is relatively weak and therefore difficult to measure even in optically active systems. The observable effect is less than 0.1%. Therefore, there is an ever-increasing demand for efficient detection and amplification of chirality. Furthermore, insight into chiral dynamics in a solvent-free environment requires chiral recognition in gas-phase, which is even more challenging due to low number densities.
-
Gas phase chiral detection techniques that have recently emerged are: (i) Photoelectron circular dichroism where the asymmetry in the angular distribution of ionized electrons is recorded, (ii) Mass spectrometry that uses enantiomer-selective binding or separation techniques, (iii) Coulomb explosion imaging that records the momentum of all the fragment ions that are produced when rapid ionization causes the molecule to explode, and (iv) chiral high harmonic generation (cHHG) with elliptically polarized light in which a slight disparity in the laser-driven electron dynamics in the two enantiomers is amplified by several orders of magnitude. The focus of this project is to develop novel techniques to study the interaction of gas-phase chiral molecules with light.
-
Moreover, all chiroptical methods to-date use the spin-angular momentum of light, that is, elliptical or circular polarization of the electromagnetic wave. However, laser beams can also be engineered to carry a well-defined orbital angular momentum (OAM). Each photon in the beam carries an orbital angular momentum of lħ. l = 0 corresponds to a Gaussian beam with a sequence of planar wave fronts. In l = 1 beam, the wave front looks like a corkscrew undergoing one full rotation in a wavelength, clockwise or counter-clockwise, resulting in helical beams (l = ±1). For l ≠ 0, OAM beams are characterized by an optical vortex along the beam axis. OAM beams may open up new selection rules during photo-ionization/excitation that have hitherto not been explored.
Photoionization with OAM beams
-
Very little is known about the role played by orbital angular momentum (OAM) of light on strong field ionization of atoms/molecules, as both experiments and theoretical calculations are sparse. The few calculations that exist involve photo-excitation of atoms showing the origin of new selection rules. When translated to molecules, it is feasible to control the molecular dynamics with the OAM of light by adding a new dimension.