Past Results
2023
St-Jacques et al. (2023) Nature Communications, 14, 6058.
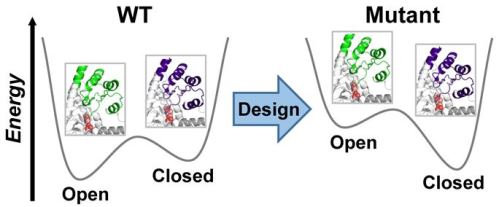
Structural plasticity of enzymes dictates their function. Yet, our ability to rationally remodel enzyme conformational landscapes to tailor catalytic properties remains limited. Here, we report a computational procedure for tuning conformational landscapes that is based on multistate design of hinge-mediated domain motions. Using this method, we redesign the conformational landscape of a natural aminotransferase to preferentially stabilize a less populated but reactive conformation and thereby increase catalytic efficiency with a non-native substrate, resulting in altered substrate selectivity. Steady-state kinetics of designed variants reveals activity increases with the non-native substrate of approximately 100-fold and selectivity switches of up to 1900-fold. Structural analyses by room-temperature X-ray crystallography and multitemperature nuclear magnetic resonance spectroscopy confirm that conformational equilibria favour the target conformation. Our computational approach opens the door to targeted alterations of conformational states and equilibria, which should facilitate the design of biocatalysts with customized activity and selectivity.
Read more here.
Legault S et al. (2022) Chemical Science, 13, 1408-1418
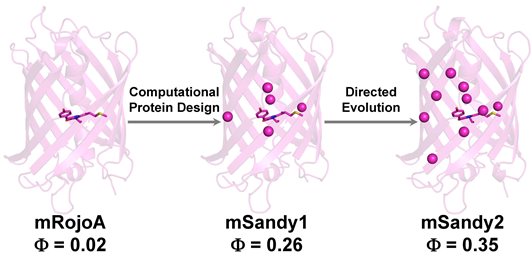
Here, we use computational protein design to enhance red fluorescent protein brightness by optimizing packing of the chromophore, which we hypothesized would increase quantum yield by rigidifying it and reducing non-radiative decay. Residues surrounding the chromophore were simultaneously mutated in silico to various combinations of aliphatic amino acids. We experimentally characterized the top 10 designed sequences and identified mSandy1, a variant displaying an 11-fold enhancement to quantum yield relative to its parent RFP (absolute quantum yield increase of 0.24), a result that has previously only been achieved using multiple rounds of directed evolution with high-throughput screening of several thousand variants. To further improve brightness, we performed three rounds of directed evolution on mSandy1 and obtained mSandy2, the brightest Discosoma sp. derived monomeric RFP with an emission maximum >600 nm reported to date. Crystallographic analysis confirmed that the chromophore p-hydroxybenzylidene moiety of mSandy2 was rigidified due to tight packing by aliphatic residues, confirming our original hypothesis. Our results demonstrate the utility of computational protein design for increasing RFP quantum yield by enhancing chromophore packing, and generating novel templates for directed evolution of brighter variants
Quote: "While many RFPs are now available, novel variants displaying high brightness in the far-red or near-infrared optical window are still desired to enable various types of applications. In this context, the [computational protein design] approach for enhancing quantum yield described here can expedite the creation of novel bright RFP templates for further engineering."
2020
Broom A et al. (2020) Nature Communications 11, 4808
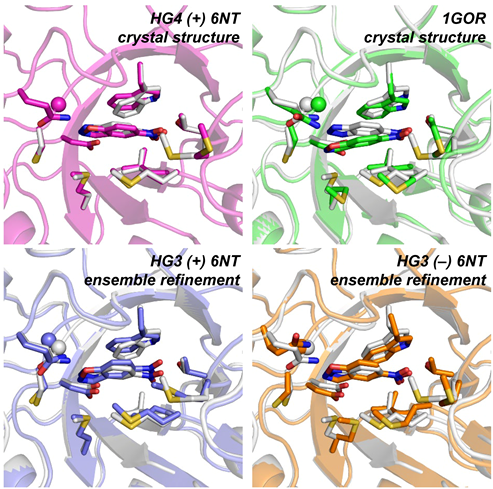
The creation of artificial enzymes is a key objective of computational protein design. Although de novo enzymes have been successfully designed, these exhibit low catalytic efficiencies, requiring directed evolution to improve activity. Here, we use room-temperature X-ray crystallography to study changes in the conformational ensemble during evolution of the designed Kemp eliminase HG3 (kcat/KM 146 M–1s–1). We observe that catalytic residues are increasingly rigidified, the active site becomes better pre-organized, and its entrance is widened. Based on these observations, we engineer HG4, an efficient biocatalyst (kcat/KM 103,000 M–1s–1) containing key first and second-shell mutations found during evolution. HG4 structures reveal that its active site is pre-organized and rigidified for efficient catalysis. Our results show how directed evolution circumvents challenges inherent to enzyme design by shifting conformational ensembles to favor catalytically-productive sub-states, and suggest improvements to the design methodology that incorporate ensemble modeling of crystallographic data.
Quote: "Computational enzyme design with a crystallographically derived backbone ensemble derived from a low-activity enzyme could obviate the need for directed evolution..."
2019
Damry AM et al. (2019) Communications Biology 2, 433
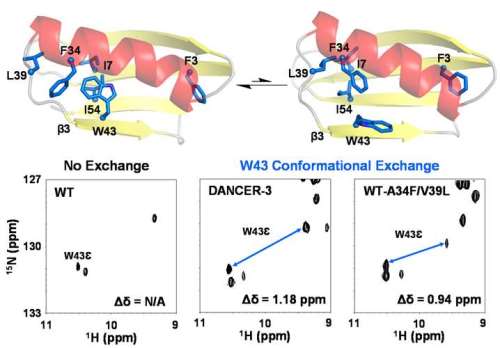
Here, we study a key question in protein biophysics: how do novel modes of conformational exchange (i.e. dynamics) arise in globular proteins. To do so, we investigated the contribution of individual mutations in DANCER-3, a pentamutant of streptococcal protein G domain β1 (Gβ1), which we previously designed to spontaneously switch between two conformations that have not been previously observed for this fold. Using a combination of solution NMR and molecular dynamics simulations, we demonstrate that only two of the five mutations are necessary to create this conformational exchange, and that these mutations work synergistically when introduced into the wild-type background, with one destabilizing the native Gβ1 structure and the other allowing two new conformational states to be accessed on the energy landscape. Based on these results, we propose a biophysical model for the molecular evolution of conformational dynamics in globular proteins in which a permissive mutation that silently reshapes unexplored regions of the protein energy landscape could spontaneously emerge, followed by a destabilizing mutation that would give rise to a new dynamic process by making the native state less stable than the alternate conformations now made accessible by the permissive mutation.
2019
St-Jacques AD et al. (2019) ACS Catalysis 9, 5480-5485
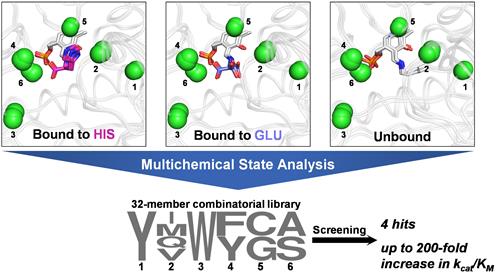
Here, we developed a multistate computational protein design methodology named multichemical state analysis (MCSA), which can be used to optimize enzyme sequences for productive binding of multiple target substrates. We used MCSA to redesign the active site of branched-chain amino acid aminotransferase to accept both α-ketoglutarate and the non-native substrate L-histidine. Screening of a designed combinatorial library comprising 32 mutants for enhanced L-histidine transamination activity yielded four variants displaying up to ≈200-fold improvements to kcat/KM with the non-native substrate (12.5% hit rate), a library size several orders of magnitude smaller than was required to obtain similar activity enhancements in other aminotransferases with a single round of directed evolution. MCSA opens the door to the rational design of broad-specificity biocatalysts and multi-substrate enzymes displaying tailored specificity.
2019
Parmeggiani F et al. (2019) ACS Catalysis 9, 3482–3486.
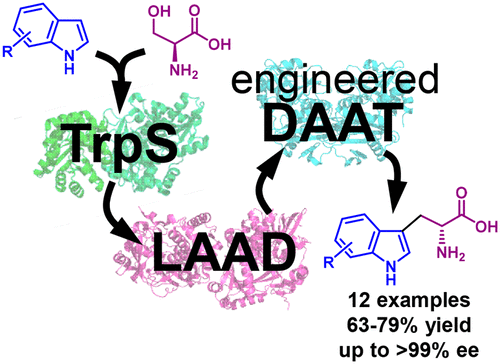
In this collaborative project with the group of Nicholas Turner (U. Manchester), we used rational design to engineer the first reported aminotransferase displaying native-like catalytic activity towards D-tryptophan (kcat/KM = 700 M-1 s-1). This engineered variant enabled us to develop a one-pot biocatalytic process that combines asymmetric synthesis of substituted L-tryptophans from indoles by tryptophan synthase (TrpS), with a stereoinversion cascade based on L-amino acid deaminase (LAAD) and our engineered aminotransferase (DAAT). We show that this biocatalytic process can be used for the synthesis of 12 D-tryptophan derivatives containing electron-donating or withdrawing substituents at all benzene-ring positions on the indole group, with high conversion rate (84% to >99%) and enantiomeric excess (91% to >99%) starting from commercially-available materials. We also demonstrate that our process is applicable to preparative-scale synthesis of all 12 D-tryptophans (isolated yields of 63% to 79%), many of which are highly-valuable building blocks of pharmaceuticals and natural products.
2017
Davey et al. (2017) Nature Chemical Biology 13, 1280-1285.
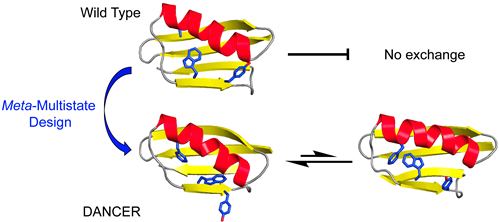
This manuscript presents the first demonstration of the rational design of proteins that can spontaneously exchange between two predefined conformational states in the absence of an external stimulus, and on a timescale relevant to function. To achieve this result, we developed a broadly-applicable computational method to engineer protein dynamics that we term meta-multistate design (meta-MSD). We used meta-MSD to design spontaneous exchange between two novel conformations introduced into the global fold of protein G domain β1 (Gβ1). The designed proteins, named DANCERs, for Dynamic And Native Conformational ExchangeRs, are stably folded and exchange between predicted conformational states on the millisecond timescale. Our new method for design of conformational exchange paves the way to the design of proteins with a more versatile range of functions than was previously possible, such as those that must adopt more than one conformational state (e.g., enzymes, molecular rotors, biosensors).
2016
Pandelieva AT et al. (2016) ACS Chemical Biology 11, 508-517.
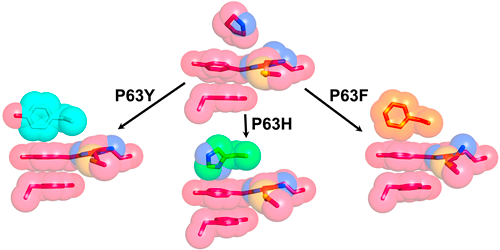
2016/09/01. We recently used rational design to increase the quantum yield and brightness of red fluorescent proteins (RFPs) by rigidifying their chromophore via the creation of a triple-decker motif of aromatic rings (see figure below). The best mutant identified displayed an over three-fold improvement relative to the parent RFP and was isolated following the screening of only 48 mutants, a library size that is several orders of magnitude smaller than those previously used to achieve equivalent gains in quantum yield in other RFPs.