|
QNANO
|
NOTE 1:
One of the most frequently used parameters is the resources directory specified by "resources_dir". This will be used to find resources like material, lattice or orbital files. A parameter of type "resource" is a string that will be interpreted as a filename. The file will be searched in the local directory, optionally appending the suffix ".dat". If no such file is present in the local directory, the file (with and optional suffix ".dat") will be looked for in the resources directory. If called from the command line, "resources_dir" should typically be "$QNANO_DIR/resources".
NOTE 2:
The main input of the QNANO framework is a position file. When applicable, it will be specified by the parameter "positions". However, whenever a position file is required, we might alternatively generate the structure on the fly using the parameter "generate_positions_from_material", which takes 4 arguments: the material name (resource), and the numbers of unit cells Nx, Ny and Nz along the 3 primitive vectors.
NOTE 3:
Some vector-type parameters are available in two variants, where one has the suffix "_LU". This indicates "lattice units": The n-th vector component is multiplied with the n-th primitive lattice vector. The lattice is either specified by a separate parameter or, if a positions file is specified that contains a reference to material files, the equilibrium lattice of the first found material is used.
In what follows, the "Types" of parameters have the meaning:
| Type Name | Meaning |
|---|---|
| Integer | A single integer number (C++: int) |
| Real | A real-valued floating point number (C++: double) |
| Boolean | A Boolean value "true" or "false" (C++: bool) |
| String | A string of characters (C++: std::string) |
| Resource | A resource file name (cf. Preliminary Notes!) |
| Vector(N) | A vector of N real-valued floating point numbers |
Furthermore, we sometimes have "List of TYPE" for some type "TYPE". This implies that multiple instances of the respetive parameter are parsed, e.g., the "List of Vector(3)" variable "periodic_vec_LU" could be invoked by
-periodic_vec_LU 1 0 0 -periodic_vec_LU 0 1 0
For "List of Strings" in particular, where there is no ambiguity, we can alternatively place the strings simply one after the other, such as in
-EV_file EV0.dat EV1.dat EV2.dat EV3.dat
This allows one to use bash-shell replacements à la
-EV_file EV[0-3].dat
Purpose: Generation of basic structures. The default is to create 1x1x1 unit cells of a given material. The number of unit cells can be adjusted and advanced options allow to cut out some predefined shapes.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| material | resource | Material file name. This file contains the information required to set up the structure of a single unit cell. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| Nx, Ny, Nz | Integer | 1 | Number of unit cells in the direction of the first, second, and third primitive vector |
| outfile | String | "positions.dat" | Name of generated positions file |
| cut_hexagon_zigzag | Integer | NOTE: only works for "hex2D" lattice (e.g., graphene)! Generates and cuts out a hexagonal flake with zigzag edges. The number specified by this parameter corresponds to the number of hexagonal benzene rings along one side of the hexagon. | |
| cut_hexagon_armchair | Integer | NOTE: only works for "hex2D" lattice (e.g., graphene)! Generates and cuts out a hexagonal flake with armchair edges. The number specified by this parameter corresponds to the number of hexagonal benzene rings along one side of the hexagon. | |
| use_center_as_origin | Boolean | false | If structures are cut (cf. cut_radius or cut_plane), the origin for cutting processes has to be defined. By default the origin is set to (0,0,0), which is the starting point from which the Nx x Ny x Nz is constructed. Setting "use_center_as_origin" to "true" will shift the origin to the center of a bounding box to the generated Nx x Ny x Nz sample. |
| cut_origin | Vector(3) | (0, 0, 0) | Set origin for cutting manually |
| cut_origin_plus | Vector(3) | (0, 0, 0) | Shift origin by adding a vector to the original origin |
| cut_radius | Real | 0. | If larger than 0: After a block of unit cells has been specified (set Nx, Ny and Nz to large enough values) all atoms outside of a sphere with radius "cut_radius" are removed |
| cut_plane | Vector(4) | Remove atoms beyond a plane specified by its Hesse normal form. Uses the "cut_origin". | |
| cut_plane_through | Vector(6) | Remove atoms beyond a plane specified by the vector perpendicular to the plane and pointing outward and a vector determining the origin explicitly. | |
| cut_plane_through_LU | Vector(6) | Remove atoms beyond a plane specified by the vector perpendicular to the plane and pointing outward and a vector determining the origin explicitly. The origin is specified in lattice units, i.e., in multiples of lattice primitive vectors. | |
| cut_plane_through_LU_LU | Vector(6) | Remove atoms beyond a plane specified by the vector perpendicular to the plane and pointing outward and a vector determining the origin explicitly. Both, the direction vector as well as the origin are specified in lattice units. |
Purpose: Takes position file and shifts all atoms by a vector.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Name of position file. |
| outfile | String | Name of output file. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| shift_nm | Vector(3) | (0., 0., 0.) | Shift vector in nm. |
| shift_LU | Vector(3) | (0., 0., 0.) | Shift vector in lattice units. |
| material | Resource | Material from which the lattice for the lattice units is deduced. If not specified, the material is taken to be the first one specified in the position file. |
Purpose: Shifts all atom positions so that the new origin in the center of a bounding box. Otherwise, using the "generate" tools, the origin is typically the point from which a regular lattice of the underlying material is constructed. This tool is particularly useful in combination with external potentials.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Name of position file |
| outfile | String | Name of generated position file |
Purpose: From samples by adding two or more positions files
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | List of Strings | Names of position files to be added |
| outfile | String | Name of generated position file |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| shift_nm | Vector(3) | (0., 0., 0.) | All position files except for the first will be shifted by the specified vector (units: nm) before being added. |
| shift_LU | Vector(3) | (0., 0., 0.) | All position files except for the first will be shifted by the specified vector (units: lattice units) before being added. |
Purpose: Substitute elements in a position file.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Name of input position file |
| outfile | String | Name of generated position file |
| replace | List of Vector(2) | Element identified by first argument is replace by the second argument |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| density | Real | 1. | Probability of atom being replaced. Used for alloying. |
| seed | Integer | 0 | Random seed for probabilistic replacement. |
Purpose: Combine two position files, where the atom types (elements) specified in the 'template' file are replaced by the types of the atoms specified in the 'substitute' file.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| template | String | Template position file with atoms to be replaced |
| substitute | String | Position file with atoms that replace the ones in the 'template' file |
| outfile | String | Name of generated position file |
Purpose: Strain relaxation
NOTE: This tool requires a version of PETSc with real scalar type to be installed on the system. However, the system default should be that $PETSC_DIR points to a PETSCc version with complex scalar type for the solution of Hermitian eigenvector problems. Here, we assume that an environment variable $PETSC_DIR_REAL is set that points to the real version of PETSc. To switch to this version, one typically has to execute the tool with an overriden LD_LIBRARY_PATH variable:
LD_LIBRARY_PATH=$PETSC_DIR_REAL/lib:$LD_LIBRARY_PATH $QNANO_DIR/bin/strain_optimize_tao ...
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Unrelaxed positions file |
| outfile | String | Output (relaxed) position file |
| resources_dir | String | Name of generated position file |
Optional parameters for PETSc Tao, e.g.:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| tao_gatol | Real | ? | Absolute error tolerance for the modulus of the gradient |
| tao_max_it | Integer | ? | Maximimum number of iterations. Should be set to a value large enough that the tolerance can be reached. |
Purpose: Generates a parabolic confinement potential in one direction: ![$ V(\mathbf{r})= a \big[(\mathbf{r}-\mathbf{r}_c)\cdot \mathbf{d}\big]^2 $](form_0.png)
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Position file containing atoms positions on which the potential is evaluated. |
| outfile | String | Output file containing the potential evaluated at the atoms from the position file. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| strength | Real | 1. | Strenght of potential 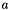 in meV/nm². in meV/nm². |
| direction | Vec3d | (0,0,0) | Direction vector 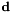 in nm. in nm. |
| center | Vec3d | (0,0,0) | Center of potential 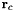 in nm. in nm. |
| truncate_radius | Real | -1. | Truncation radius. If larger than zero, the parabolic potential is capped at 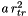 when when 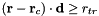 |
| add_to | String | Adds generated potential to an existing potential from file specified by this parameter. |
Purpose: Generates a parabolic confinement potential in one direction: ![$ V(\mathbf{r})= a \big[(\mathbf{r}-\mathbf{r}_c)^2- \big((\mathbf{r}-\mathbf{r}_c)\cdot \mathbf{d}\big)^2\big] $](form_6.png)
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Position file containing atoms positions on which the potential is evaluated. |
| outfile | String | Output file containing the potential evaluated at the atoms from the position file. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| strength | Real | 1. | Strenght of potential in meV/nm². |
| direction | Vec3d | (0,0,0) | Direction vector in nm. |
| center | Vec3d | (0,0,0) | Center of potential in nm. |
| truncate_radius | Real | -1. | Truncation radius. If larger than zero, the parabolic potential is capped at when |
| add_to | String | Adds generated potential to an existing potential from file specified by this parameter. | |
| set_truncate_offset_zero | Boolean | false | After truncation: shift potential so that the potential cap is set to zero. |
Purpose: Generate potential file describing a linear potential discretized on the atoms of a given sample: 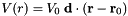 .
.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Name of input position file |
| outfile | String | Name of generated position file |
| strength or strength_LU | Real | strength  of the potential in [eV/nm] or [eV] if lattice units are used. of the potential in [eV/nm] or [eV] if lattice units are used. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| center | Vector(3) | (0, 0, 0) | Center  of the coordinate system. of the coordinate system. |
| direction | Vector(3) | (0, 0, 1) | Direction , will be normalized. If "strength_LU" is specified, the direction is interpreted in lattice units before normalization. |
| add_to | String | After calculation, the potential is added to the potential from the file specified by this parameter. |
Purpose: Truncates potential: every value of the potential above (or below) a given threshold is set to the threshold value.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| potential | String | Name of the input potential file. |
| outfile | String | Name of the output potential file. |
| threshold | Real | Threshold at which the potential is capped |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| from_below | Boolean | false | Set to true if potential is to be capped from below instead of from above. |
Purpose: Adds two or more potential files.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| potential | String | Basic potential file to which others are added. |
| add | List of Strings | Names of other potential files. |
| outfile | String | Output file containing the summed potential. |
TODO: EXPAND
Purpose: Display structures by generating postscript files. The output can be opened with a standard ps/pdf reader like "evince" on Linux or it can be converted by most image processing tools to another graphics format.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| positions | String | Name of positions file of the structure to be depicted |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| outfile | String | "QNANO2ps.ps" | Name of the generated postscript file. Should end in ".ps" |
| displayEV | List of Strings | Name of a single-particle eigenvector file. When used, the charge density of the eigenstate will be plotted. If multiple file names are specified, the charge densities will be added (useful, e.g., for degenerate states). | |
| potential | String | Name of a potential file. When used, the potential on the selected atoms will be plotted. | |
| lookfrom | "x", "y" or "z" | "z" | Direction from which the structure is depicted |
| quaternion | Vector(4) | (1, 0, 0, 0) | Direction from which the structure is depicted, defined by a quaternion |
| boxwidthx, boxwidthy, boxwidthz | Real | 0 | Used if > 0: Plotting the structure from one side implies that everything below the surface will be covered by the surface atoms. We can filter out a subset of atoms to be displayed by defining a box (width in nm) around the center of the sample. For example, when looking from the z-direction, setting "boxwidthz" to a small value ~0.3 allows us to depict a cross section though the center of the sample. |
| translatex, translatey, translatez | Real | 0 | Translates the center of box of selected atoms. |
| atom_radius | Real | 0.05 | Radius of the circles depicting the atoms (in nm). |
| atom_border_width | Real | 0.001 | By default the circles depicting the atoms have a small gray border. This parameter defines its size in nm. Set to 0 if you don't want a border on the atoms. |
| postscript_nm_to_pt | Real | 30. | Scale with which distances in nm will be converted into pt in the postscript. |
Purpose: SLEPc-based solver for single-particle tight-binding Hamiltonians. MPI-Parallelized, so try calling by $MPIRUN -n $NTHREADS $QNANO_DIR/bin/tightbinding_slepc.
USES ALL PARAMETERS OF: TB_Model_Slater_Koster and Eigenvector_Printer as well as additional SLEPc/PETSc command line options
Typical useful SLEPc options:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| eps_target | Real | 0. | Target value: Defines spectral region where eigenstates are looked for. Furthermore, eigenvalues/eigenvectors will be printed in ascending order of the absolute difference to the target value. |
| eps_nev | Integer | 1. | Number of eigenstates to be calculated. |
| eps_monitor, eps_monitor_all | No Argument | Prints information after each iteration step. | |
| eps_harmonic | No Argument | Turns on harmonic extraction. This can speed up the convergence significantly. However, know what you are doing an look it up in the documentation for SLEPc. If harmonic extraction is used, you should also set "eps_ncv" to an integer at least twice the size of "eps_nev", but not too large because the memory demands will increase as well. | without_strain_correction | No Argument | false | Should be set to true for all cases! |
Purpose: Exact solver for single-particle tight-binding Hamiltonians using Eigen library.
USES ALL PARAMETERS OF: TB_Model_Slater_Koster and Eigenvector_Printer
Additional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| eps_target | Real | 0. | Target value: Defines spectral region where eigenstates are looked for. Furthermore, eigenvalues/eigenvectors will be printed in ascending order of the absolute difference to the target value. |
Purpose: Band structure calculation tool. More precisely, caluculates tight-binding problems while multiplying each hopping element by 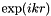 for a number of values for
for a number of values for 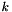 , where
, where 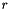 is the distance vector between the respective neighbors.
is the distance vector between the respective neighbors.
USES ALL PARAMETERS OF: TB_Model_Slater_Koster and Eigenvector_Printer
Additional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| kpt_file | String | required if "kpt_path_point" not set | k-point file in the same format as in QUANTUM ESPRESSO |
| kpt_path_point | Vector(4) | required if "kpt_file" not set | First 3 elements: k-point in units of reciprocal lattice vector (single unit cell specified by lattice file). 4th element: Number of discretization points from this k-point to the next specified point (exclusive). C.f. k-point path format in QUANTUM ESPRESSO. |
| scale_k | Vector(3) | (1., 1., 1.) | Scales the first 3 numbers of 'kpt_path_point' (or of each line in the 'kpt_file') by the respective scaling factor specified here. Useful for periodic structures with more than 1x1x1 unit cells. For example, for a 2x2x1 unit cell, set scale_k to (0.5, 0.5, 1). |
NOTE: "-periodic_vec_LU" from TB_Model_Slater_Koster for periodic structures is not automatically set! Do so manually!
Purpose: Show some information about single-particle eigenstates. Useful for sanity checks.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| resources_dir | String | Resources directory, usually "$QNANO_DIR/resources" |
| positions | String | Name of positions file of the structure to be depicted |
| EV_file | List of Strings | Names of single-particle eigenvector files. |
Purpose: Print complex phase of the local orbital expansion coefficient of single-particle eigenvector files with maximal density.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| EV_file | List of Strings | Names of single-particle eigenvector files. |
Purpose: Calculates the scalar product between one single-particle eigenstate and a number of others
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| EV_file | List of Strings | Names of single-particle eigenvector files. The scalar product of the first state in the list with all the others will be calculated. At least two files have to be specified. |
Purpose: Calculate spins of single-particle states. Used as input for the tool $QNANO_DIR/bin/manybody_spins.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| resources_dir | String | Resources directory. Usually "$QNANO_DIR/resources". |
| outfile | String | Name of output file. |
| nr_e and/or nr_h | Integer | Number of electron or hole states. |
| EV_filename_pattern_e and/or EV_filename_pattern_h | String | Pattern for conduction band electron (_e) or valence band electron (_h) single-particle eigenvector files. Has to contain a '*', which is then replaced by a number ![$i \in [0,1,\dots,n_{e/h}] $](form_13.png) for a concrete eigenstate. for a concrete eigenstate. |
Purpose: Calculate dipole matrix elements between a set of single-particle states. Currently, only shift-terms are calculated.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| resources_dir | String | Resources directory. Usually "$QNANO_DIR/resources". |
| positions | String | Name of positions file. |
| outfile | String | Name of output file. |
| nr_e and/or nr_h | Integer | Number of electron or hole states. |
| EV_filename_pattern_e and/or EV_filename_pattern_h | String | Pattern for conduction band electron (_e) or valence band electron (_h) single-particle eigenvector files. Has to contain a '*', which is then replaced by a number for a concrete eigenstate. |
TODO: EXPAND
Purpose: Pre-computation of onsite Coulomb matrix element for a given element (only done once for a new material).
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| orbfile | String | Name of the orbital file specifying the Slater orbitals and the orbitals to be used. |
| outfile | String | Name of the output file for the precomputed onsite Coulomb matrix elements. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| lim | Real | 2.5 | Integration limits: -lim ... lim |
| Ndiscr | Integer | 1e8 | Number of sample points for the VEGAS algorithm. |
Purpose: Calculation of Coulomb matrix elements between selected single-particle eigenstates. MPI-Parallelized, so try calling by $MPIRUN -n $NTHREADS $QNANO_DIR/bin/coulomb.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| resources_dir | String | Resources directory. Usually "$QNANO_DIR/resources". |
| positions | String | Name of the positions file. |
| dielectric_constant | Real | Dielectric constant for long-range terms. Only required if "no_longrange" is "false". |
| nr_e, nr_h, nr_e_LR, nr_h_LR, nr_e_OS and/or nr_h_OS | Integer | Number of electron (nr_e...) or hole (nr_h...) states. Can be set separately for long-range (LR) and onsite (OS) terms. |
| EV_filename_pattern_e and/or EV_filename_pattern_h | String | Pattern for conduction band electron (_e) or valence band electron (_h) single-particle eigenvector files. Has to contain a '*', which is then replaced by a number for a concrete eigenstate. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| outfile_direct | String | "Vdirect.dat" | Name of output file for direct electron-hole terms |
| outfile_exchange | String | "Vexchange.dat" | Name of output file for exchange electron-hole terms |
| outfile_ee | String | "Vee.dat" | Name of output file for electron-electron terms |
| outfile_hh | String | "Vhh.dat" | Name of output file for hole-hole terms |
| nr_blocks | Integer | 1 | Block decomposition of matrix-vector products for long-range terms. Set to larger value if program takes too much memory. This, however, comes at the cost of increasing overhead and therefore computation time. |
| no_dir, no_exc, no_ee, no_hh | Boolean | false | Used to turn off calculation of direct, exchange, electron-electron or hole-hole terms |
| no_longrange, no_onsite | Boolean | false | Used to turn off calculation of long-range or onsite terms |
| onsite_coulomb | List of Pairs(String, String) | Load precomputed onsite Coulomb matrix element for a specific material. First argument per pair is an orbital file, the second argument is the file containing the precomputed onsite Coulomb matrix elements. |
SAME AS FOR $QNANO_DIR/bin/manybody_eigen plus additional SLEPc/PETSc command line options!
USES ALL PARAMETERS OF: Eigenvector_Printer
Additional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| rank_e, rank_h | Integer | Required | Rank, i.e., number of electron and hole in the excitonic complex, e.g, rank_e=1 and rank_h=2 for positive trions. |
| nr_e, nr_h | Integer | Required | Number of electron and hole single particle states from which the basis of Slater determinants is constructed. |
| cb_energies, vb_energies | String | Required | Name of files containing conduction and valence band single-particle energy eigenvalues. |
| Vdir, Vexc, Vee, Vhh | String | Coulomb matrix element files for direct, exchange, electron-electron and hole-hole terms. | |
| no_dir, no_exc, no_ee, no_hh | Boolean | false | Used to turn off direct, exchange, electron-electron or hole-hole states. |
| manybody_hilbertspace | String | "" | If "" or "CI", the full CI is calculated within the selected Hilbert space (cf. select_nr_e/select_nr_h/select_start_e/select_start_h). Other values: "CIS": CI + single excitations (up to nr_e/nr_h single-particle states), "CISD": CI + single and double excitations, "CISDT": CI + up to triple excitations, "CISDTQ": CI + up to four excitations. |
| select_nr_e, select_nr_h | Integer | nr_e, nr_h | If specified, the basis of Slater determinants is created up to select_nr_e/select_nr_h instead of nr_e/nr_h. This multi-configurational basis can be used as a starting point for calculations with a reduced Hilbert space, e.g., CIS/CISD/... (Configuration-interaction + single & double excitations). nr_e and nr_h then serve determine the respective full CI Hilbert space to be approximated. |
| select_start_e, select_start_h | Integer | 0 | Not all single-particle states are used, select_start_e/select_start_h states will be skipped. |
| print_selected_states | Boolean | false | If set to true, a list of selected states (reduced Hilbert space) is printed to the file "TEST_SELECTED_STATES.dat" |
| add_selected_states | String | "" | States listed in this file are added to the Hilbert space. |
| PT_outfile | String | "" | If specified, perturbative calculation will be performed in a larger Hilbert space with up to "nr_e_PT" and "nr_h_PT" single-particle states. |
| nr_e_PT, nr_h_PT | Integer | 0 | C.f. parameter "PT_outfile" |
| threshold | Real | 0 | If set, a single step of a perturbative selected-state CI algorithm is performed. New states for the next step are selected when their perturbative weight exceeds the threshold. |
| new_states_file | String | "" | Used if "threshold" is set: Specifies the name of the file in which the newly selected states are printed. |
| add_new_states_file | String | "" | Used if "threshold" is set: Specifies the name of the file in which the newly selected states are printed. New states will be printed in addition to the states already existing in the file. |
| small_denominator_threshold | Real | 1e-5 | If "PT_outfile" and "threshold" are specified: also select states when the energy denominator is smaller than this value. |
TODO: EXPAND
Purpose: Extracts dipoles and transition energies from high-rank many-body states with n_e electrons and n_h holes to low-rank many-body states with n_e-1 electrons and n_h-1 holes.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| dipole_file | String | Name of the single-particle dipole file, typically generated by $QNANO_DIR/bin/dipole. |
| outfile | String | Name of output file. |
| high_rank_EV_pattern | String | Patterns of many-body eigenvector files describing the high-rank complexes, i.e., the initial complexes for emission processes. Has to contain an "*". |
| low_rank_EV_pattern | String | Patterns of many-body eigenvector files describing the low-rank complexes, i.e., the final complexes for emission processes. Has to contain an "*". Can be empty if high-rank files describe excitons. |
| nr_high_rank / nr_low_rank | Integer | Number of high-rank and low-rank eigenvector files to be read. nr_low_rank only needs to be set if the high-rank eigenvectors do not correspond to excitons. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| high_rank_PT_file/low_rank_PT_file | String | "" | If many-body calculations are performed with perturbative corrections, the many-body eigenvector files contain the unperturbed eigenvalues. Specify the file names set by the parameter "PT_outfile" of $QNANO_DIR/bin/manybody_eigen or $QNANO_DIR/bin/manybody_slepc to add perturbative corrections when calculating spectra. |
Purpose: Used to plot an emission spectrum with delta-functions broadened to Lorentzians and initial states occupied thermally.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| spectra | String | Name of the input spectra file, typically generated by $QNANO_DIR/bin/spectra. |
| outfile | String | Name of output file. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| linewidth | Integer | 1e-4 | Width of the spectral lines (Lorentzians) in eV. |
| temperature | double | 0 | Temperature (in K) describing the occupation of initial states. |
| Estart, Eend | double | calculated from "spectra" | Energy range in which the spectrum is plotted. Default: lowest to highest energy in the "spectra" file. |
| Ndiscr | Integer | 0 | Number of sample intervals of the energy range. Default: calculated from Eend, Estart and linewidth. |
Purpose: Structure to set up general tight-binding Hamiltonians with Slater-Koster parametrization.
Required parameters:
| Name | Type | Meaning |
|---|---|---|
| resources_dir | String | Resources directory. Usually "$QNANO_DIR/resources". |
| positions | String | Name of the positions file. Only required if "generate_positions_from_material" is set. |
| generate_positions_from_material | String+Vector(3) | Instead of supplying a positions file, we can also generate Nx x Ny x Nz unit cells (arguments 2, 3, and 4) of a given material (first argument) on the fly. Not required if "positions" is specified. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| without_spinorbit | Boolean | false | Used to force turning off spin-orbit coupling. |
| without_strain_correction | Boolean | false | Used to force turning off strain corrections in tight-binding model. |
| periodic_vec_LU | List of Vector(3) | Periodicity vector for periodic structures in lattice units, i.e. smallest vector  along a certain direction for which the atom at along a certain direction for which the atom at 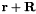 corresponds to the one at corresponds to the one at 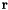 . . | |
| passivate_sp | Boolean | false | Used to turn on surface passivation assuming sp3-hybridized orbitals. |
| potential | String | Name of file containing an external potential evaluated for the same structure as provided by the "positions" parameter. | |
| no_nn2 | Boolean | false | Used to turn off second-nearest neighbors, even if they are specified in the material files. |
| evaluate_at_k | Vector(3) | - | Evaluate Hamiltonian at the specified k-point. |
| evaluate_at_k_LU | Vector(3) | - | Evaluate Hamiltonian at the specified k-point. Specified in lattice units. |
Optional parameters:
| Name | Type | Default Value | Meaning |
|---|---|---|---|
| EV_filename_pattern | String | "EV*.dat" | Pattern for printed eigenvector file name. Has to contain an '*'. |
| eigenvalue_filename | String | "eigenvalues.dat" | File name for printed eigenvalues. |
| nr_print | Integer | 0 | Number of printed eigenvectors. If <0, all converged states are printed. |
| print_Kramers_sz | Boolean | false | Used to switch on Kramers rotation: For calculations with spin but without magnetic field, time-reversal symmetry ensures that all energy eigenstates come in pairs. If "print_Kramers_sz" is set to true, those linear combinations of the two states in one pair are printed that minimize/maximize the z-component of the spin. For calculations without spin-orbit coupling, this means that the first state in each pair is a spin-down state, the second is a spin-up state. |
| kramers_tolerance | Real | 1e-7 | Determines when two are assumed to be degenerate for Kramers rotation. |
 1.8.11
1.8.11

