Research
Our group has broad interests spanning main group and transition metal chemistry. This provides a unique and stimulating learning and research environment. Discovery of new reactivity, fundamental concepts and cool chemistry is at the heart of our efforts.
Creativity, independence and breadth of interests are features that are bred and rewards that come to students and researchers in our lab.
TYPICAL/CURRENT PROJECTS
Photocatalytic and Electrocatalytic Redox Reactions
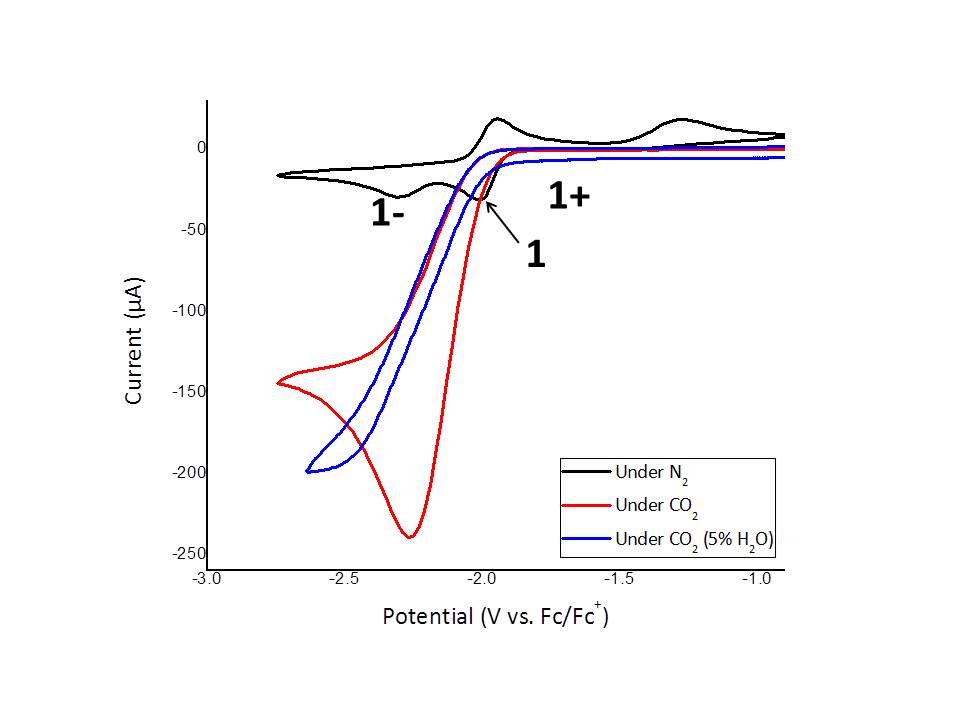
With carbon in its most oxidized state, the catalytic reduction of CO2, to yield C1 products presents a fundamental challenge in chemistry. Significant incentives for targeting this undertaking are the environmental concerns over release of CO2 into the atmosphere combined with the potential to recycle this ubiquitous combustion byproduct into energy containing products. One of our goals is to develop distinctive metal coordination geometries for reduction catalysts and we anticipated this may lead to novel reactivity. These projects will refine a student's skills in synthetic chemistry and enhance their portoflio of analytical techniques. Some of these complexes are proficient at H2 evolution. Recently we have been exploring the the combinatoin of these complexes with innovative photosensitizers to yield new photochemical redox systems.
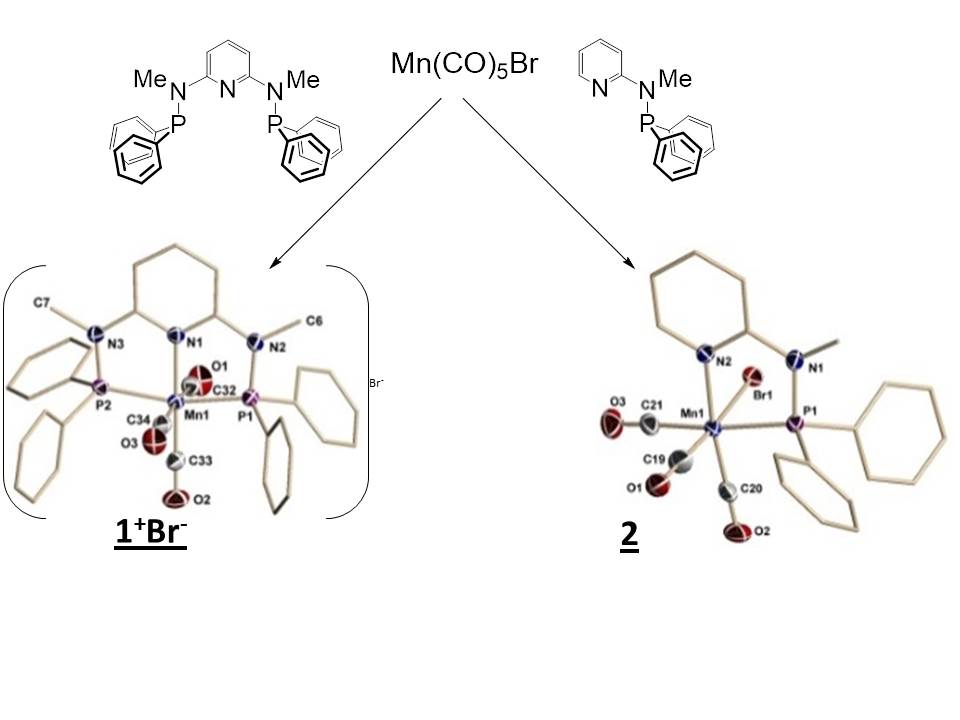
Employing pi-Conjugated Ligand Frameworks for Notable Structures and Reactivity
We have developed a number of pi-conjugated ligand scaffolds that display monoanionic or dianionic charges and coordinate in 4 to 6-membered ring motifs. Our group is a pioneer in the application of monoanionic, small bite-angle guanidinate ligands to both transition and main group metals. What began from a fundamental base blossomed into the application of these species in catalysis. We reported the first examples of transition metal catalyzed guanylation of amines with carbodiimide. The precatalysts for these reactions are novel guanidinate-supported group 4 imido species. These reactions represent a new, efficient, catalytic method for assembling guanidines from a diverse set of amines. These complexes are also capable of catalyzing the first examples of guanidine transamination thus allowing the conversion of trialkylguanidines into aryldialkylguanidines. We have expanded this effort to show that main group catalyst using Li and Al are efficient for guanylations and cynamide oligomerization.
Our application of N, N'-disubstituted 1,8-diaminonaphthalene as ligands remains a significant contribution of our group efforts. We have demonstrated the versatility of these dianionic ligands that form six-membered chelating rings in terms of application to main group and transition metal chemistry and we have expanded significantly on the variety of ligand architectures. More recently, we reported that these ligands provide a flexible scaffold and that the metal-ligand bonding interactions are more versatile than might be initially anticipated. We also expanded this framework to include a new architecture; the di(indolyl)methane dianion.
An original success that has arisen out of this area was our design and preparation of a novel carbene with an entirely new molecular architecture and electronic environment. These carbenes provide both an hospitable steric environment and the electronic activation required to generate the coordinatively unsaturated metal compounds that are vital intermediates in catalysis. We believe that this is a significant contribution to a new, exciting and growing field of research, and thus is of general interest to a wide cross-section of the chemical community.
Challenging Fundamental Concepts in Metal-Ligand Bonding
This work is an evolution of our primary interest in the design and assembly of molecules with bonding features that challenge conventional views. In particular, we are interested in noncovalent metal/ligand interactions. These concepts are under appreciated in the literature. Once revealed, these features can be extended by us to influence the physical, chemical and magnetic properties of an array of different complexes. Computational analyses continue to take an increasing role and have provided critical insight into analysis of noncovalent bonds and revealed some unanticipated molecular interactions. Our recent focus is on neutral pincer ligands coordinated to cations that have weakly coordinating anions. Most of this effort has been with a systematic substituent variation with a series of bis(imino)pyridine ligands which present a planar and orthogonal disposition of the donor orbitals. Although at first glance the ligand is oriented for the conventional bonding, structural analysis suggested otherwise and computations revealed that the metal-ligand interactions involve very little conventional donor-acceptor bonding with only 0.12-0.14 electrons being transferred from the ligand to the metal center. The corresponding bond orders are only 0.08-0.28, values substantially lower than expected for a single bond. These puzzling bonding issues continue to inspire the synthesis of increasingly challenging target complexes.
Rational Manipulation and Control of Anisoptropy of Transition Metal Complexes
This interest evolved from our explorations in main group chemistry and the challenges that this work provided to the concepts of metal-ligand covalency. One objective is to produce monometallic cobalt and iron complexes whose magnetic/electronic anisotropies reveal themselves as individual molecular magnets (so-called “single-molecule magnets” or SMMs). The fundamental goal is to understand and control the magnetic anisotropy of first row metal ions and thus remove some of the serendipity that is normally used in the synthetic work on SMMs. We are manipulating the geometry and electronic structure of five-coordinate transition metal complexes with particular focus on d7 Co(II) and d6 Fe(II) compounds. These complexes display slow relaxation of the magnetization that is characteristic SMM behavior. Through peripheral ligand modifications it is possible to modulate the orientation of the basal plane of this geometry and adjust the coordination environment of the M(II) ion. This type of structural fine-tuning can be utilized to shed light on the intricate role of spin-orbit coupling in slow relaxation of the magnetization and on designing new complexes with novel magnetic properties.