Time-Resolved Photoelectron Spectroscopy (TRPES)
The advent of commercially available, broadly tunable femtosecond laser systems over the last few years has enabled the study of molecular dynamics in real time as an initially prepared state, or set of states, evolves towards a final set of products. This is depicted schematically in figure 1. The typical timescale of such dynamical evolution in molecules occurs on the order of tens to hundreds of femtoseconds (1 femtosecond = 10-15 seconds). Using so-called ‘ultrafast’ laser pulses that are similar in temporal duration to these processes therefore allows us to follow dynamical processes in molecules as they happen.
Using photoionization as a probe to follow this dynamical evolution is an ideal tool since in principle all participating molecular states may be observed directly. This is in contrast to techniques such photoabsorption or photoemission spectroscopy, where certain ‘dark states’ may be unobservable, and the overall details of the dynamics are therefore partially obscured.
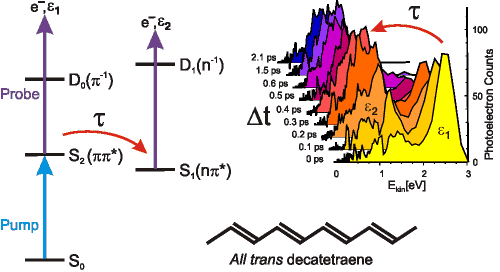
In contrast to many groups utilizing photoionization as a dynamical probe, we choose to look at the distribution of photoelectron kinetic energies rather than simply the overall photoion yield. This often provides far more subtle insight into the dynamics, as illustrated in figure 2 for the case of the molecule all trans decatetraene. The photoelectron spectrum is clearly seen to evolve as function of the delay time between the pump laser pulse (which prepares the initial state) and the probe laser pulse (which probes the evolution towards the final states at each timeste Δp,t). This is a consequence of Koopmann’s correlations: The initially prepared S2 state ionizes to the ground electronic state of the cation (termed D0), whereas the S1 state into which it evolves ionizes to the first excited electronic cation state (D1). The distribution of photoelectron kinetic energies resulting from each state are therefore markedly different, as can be seen in the figure. An important point to note is that integration over all photoelectron energies at each time step (equivalent to monitoring the photoion yield) obscures this dynamical information. TRPES is therefore a powerful tool for studying molecular dynamics.
Further information on the ionization dynamics can be obtained by looking at the photoelectron angular distribution.
Alignment
Molecules possess many properties with angular dependence (polarizability, moment of inertia, electron density, etc.) and much of this information is lost when studying an ensemble of randomly oriented gas-phase molecules. Instead, we study molecular dynamics from within a reference frame attached to (and rotating with) the molecule – the molecular frame. One way to do this is by aligning the molecules in the lab frame by applying a strong laser field. This field must be removed before probing the system since it alters the molecular properties being studied, so the alignment only lasts for a short time. Aligning molecules in this way is viable only for simple molecules (diatomic, linear) but is very difficult when three distinct axes must be aligned. For these more complex molecules, we will align only one axis and observe the molecular ensemble as it tumbles through various orientations. This information will then used to reconstruct the molecular-frame information.
he observables we use to study molecular dynamics are the photoelectron spectrum (PES) and photoelectron angular distribution (PAD), which we obtain using Velocity Map Imaging (VMI). Conventional VMI obtains a 2D projection of the 3D PAD and the 3D PAD is then recovered using an inverse Abel transform. This process is only possible when the PAD possesses cylindrical symmetry with respect to the detector axis, a property that is not present in the tumbling molecular ensemble described above. For this reason, we require a fully 3D VMI technique which does not require an inverse Abel transform. We are currently developing such a technique.