Quantum Theory Group , University of Ottawa
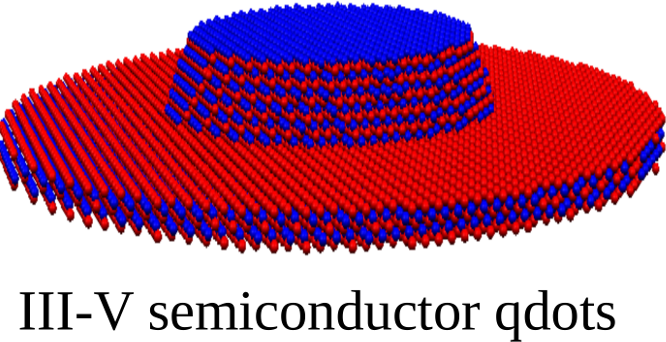
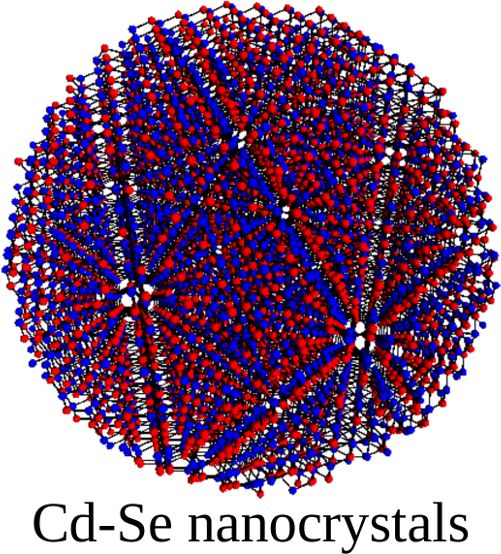

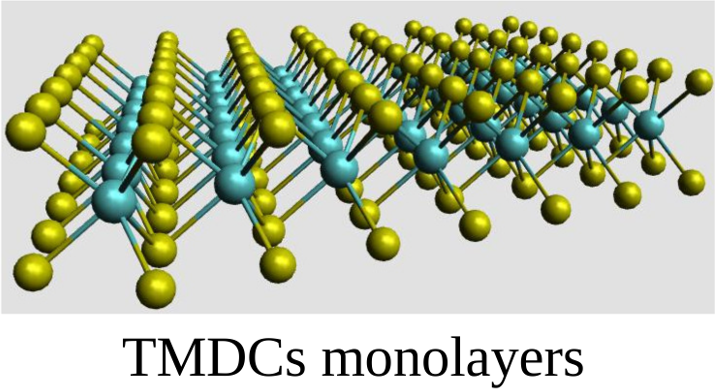
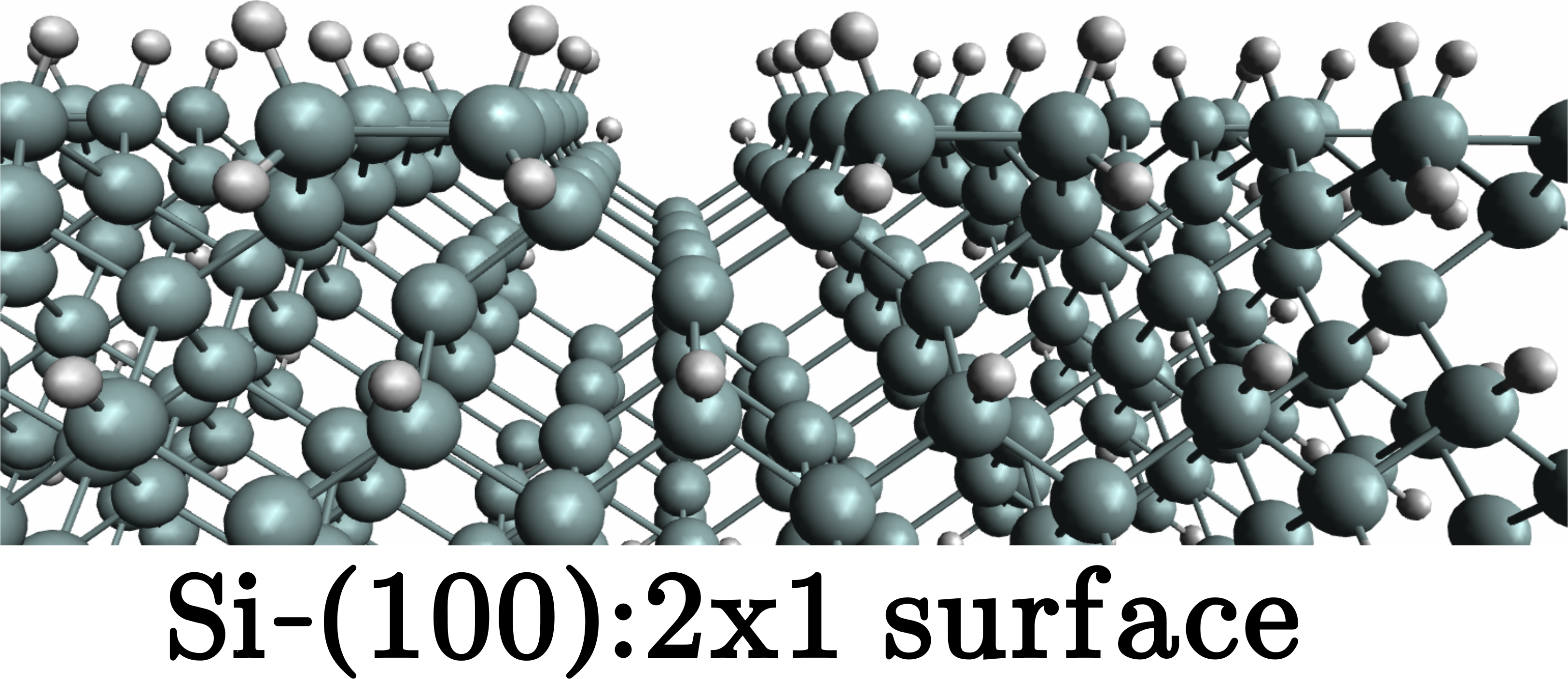
QNANO is a computational platform for the predictive simulation of the electronic and optical properties of million atom semiconductor and graphene nanostructures. QNANO starts with atomistic definition of the nanostructure, e.g., InAs quantum dot in GaAS or InP or graphene quantum dot. Different materials result in long range strain. Using in-house conjugated gradient methods the total elastic energy for up to billion atom nanostructures is minimized and equillibrium atomic positions obtained. The single-particle wavefunctions for up to a million atoms are expanded as a linear combination of atomic s, p3, d5 and s* orbitals (LCAO). The expansion coefficients are obtained by diagonalizing a parametrized tight-binding (TB) Hamiltonian for up to a million atoms. The parametrization involves ab-initio and TB calculation of the electronic bandstructure corresponding to atomic positions in strained material. The single particle levels are used as input to the atomistic calculation of Coulomb and dipole matrix elements. Hartree-Fock and DFT methods coupled with Configuration Interaction are used to compute exciton, charged exciton and multi-exciton emission and absorption spectra, Auger scattering and transport coefficients.
The underlying lattice structure, symmetry, strain and disorder in investigated nanostructures are self-contained in QNANO. Different predefined shapes of the model nanostructures, self-assembled quantum dots, nanocrystals and graphene qdots are available. The optimized numerical algorithms for elastic energy minimization, genetic algorithms for bandstructure fitting, diagonalization of large matrices, evaluation of electron-electron Coulomb matrix elements and efficient exact diagonalization tools are at the core of the QNANO.
Last modified: